Sx de Seckel
Es una enfermedad congénita (que está presente desde el nacimiento) muy rara, caracterizada por retraso del crecimiento intrauterino, microcefalia (cabeza anormalmente pequeña), dwarfismo (forma de enanismo proporcionado) y forma de cara peculiar.

Fue descrita por primera vez como "enanismo de cabeza de pájaro" por Rudolf Virchow en 1892, posteriormente en 1960 Helmunt Seckel caracterizó el síndrome tal como lo conocemos en la actualidad. Se
estima una frecuencia menor de 1/10.000 recién nacidos vivos, afectando por igual a mujeres como hombres, sin que parezca presentar predominio étnico ni geográfico.
Clínicamente se caracteriza por dwarfismo, debido a que el retraso del crecimiento intrauterino continúa después del nacimiento, rasgos cráneofaciales característicos son:

-Microcefalia
-Nariz picuda y prominente conocida como "pico-corno"
-Ojos anormalmente grandes
-Cara estrecha
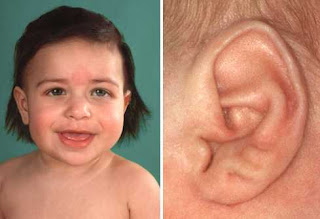
-Orejas malformadas con ausencia de lóbulo
-Paladar ojival (paladar en forma de bóveda)
-Micrognatia (mandíbula inusualmente pequeña).
Las anomalías de las extremidades son:
-Clinodactilia (arqueamiento permanente del quinto dedo) o microdactilia (pequeñez anormal de uno o más dedos debida a detención del desarrollo o ausencia congénita de alguna falange) sobre todo del 5º dedo y otras anomalías óseas.
-Se acompaña de grados variables de retraso mental.
-Menos frecuentemente se acompaña de craneosinostosis (cierre prematuro de los huesos del cráneo)
-Asimetría facial, fisuras palpebrales antimongoloides (el canto externo del ojo más bajo que el canto interno), estrabismo (desviación de uno de los ojos de su dirección normal, por lo que los ejes visuales no pueden dirigirse en un mismo punto)
-Malposición dental
-Hipoplasia (desarrollo incompleto o defectuoso) del esmalte dental
-Retraso de la maduración ósea
-Hipoplasia de los huesos del carpo
-Luxación radial (dislocación de uno de los huesos del antebrazo)
-Displasia (malformación) de caderas
-Escoliosis (curvatura oblicua anormal de la columna dorsal)
-Pliegue simiesco (un solo pliegue, profundo de las palmas de las manos)
-Anomalías de los dermatoglifos (dibujos formados por las crestas y surcos de las manos y los pies)
-Criptorquidia (en el caso de los hombres)-Hirsutismo (aumento excesivo del pelo en la mujer)
-Hipertrofia (desarrollo exagerado de un órgano, del clítoris), en la mujer.
El diagnóstico de la enfermedad es fundamentalmente clínico; el estudio radiológico puede constribuir al diagnóstico ya que permite ver las alteraciones óseas que acompañan a este síndrome.
El diagnóstivo prenatal mediante ecografía permite observar algunas de las malformaciones características del síndrome como la microcefalia y el retraso del crecimiento intrauterino, habitualmente severo, siendo más difícil visualizar las anomalías faciales. Los antecedentes familiares refuerzan la sospecha diagnóstica.
El diagnóstico diferencial debe realizarse con el Síndrome de Dubowitz, fetopatía alcohólica, trisomía 18, Síndrome de Cornelia de Lange, Síndrome de Bloom, anemia de Fanconi y otros enanismos primarios.
No existe un tratamiento curativo especifico para de la enfermedad. Por lo que sólo se puede hacer tratamiento sintomático.
El síndrome de Seckel parece heredarse como un rasgo genético autosómico recesivo.
PRESENTACIÓN DEL CASO
Se trata de un recién nacido de madre primigesta de 19 años, con embarazo de término, 3 CPN, sin patología materna reportada. El parto fue vaginal, nació cefálico, de sexo femenino, líquido amniótico teñido de meconio. Recibe un Apgar 8-9, 38 semanas de edad gestacional. Peso: 1,380 g; talla: 39 cm; PC: 26 cm. Con buen estado general y signos vitales estables, pero dismórfico, con microcefalia, inserción baja de orejas, facies anormal (Figuras 1, 2 y 3).
Se trata de un recién nacido de madre primigesta de 19 años, con embarazo de término, 3 CPN, sin patología materna reportada. El parto fue vaginal, nació cefálico, de sexo femenino, líquido amniótico teñido de meconio. Recibe un Apgar 8-9, 38 semanas de edad gestacional. Peso: 1,380 g; talla: 39 cm; PC: 26 cm. Con buen estado general y signos vitales estables, pero dismórfico, con microcefalia, inserción baja de orejas, facies anormal (Figuras 1, 2 y 3).



Fue ingresado inmediatamente en la unidad neonatal como RN de término, pequeño para la edad gestacional, RCIU severo, MBPN, a descartar una cromosomopatía. Anomalías craneofaciales: microcefalia severa, nariz picuda y prominente, ojos grandes, micrognatia y orejas de implantación bajas. Extremidades superiores: contracturas, antebrazos en flexión y encurvados, muñecas flexionadas (flexión palmar), manos deformadas, pliegue simiesco, clinodactilia. Un USG transfontanelar mostró una dilatación de los ventrículos laterales y ligera dilatación del III ventrículo.
En conclusión: ventriculomegalia vs. hidrocefalia, y se recomendó realizar una TAC craneal.
EVOLUCIÓN: El síndrome de Seckel se sospechó al tercer día de vida. Superó una neumonía de inicio temprano con antibioticoterapia. Mantuvo una adecuada alimentación enteral a través de SOG. Abandonó a pedido de la familia a la edad de 24 días (Figuras 4 y 5).
En conclusión: ventriculomegalia vs. hidrocefalia, y se recomendó realizar una TAC craneal.
EVOLUCIÓN: El síndrome de Seckel se sospechó al tercer día de vida. Superó una neumonía de inicio temprano con antibioticoterapia. Mantuvo una adecuada alimentación enteral a través de SOG. Abandonó a pedido de la familia a la edad de 24 días (Figuras 4 y 5).


El síndrome de Seckel (SCKL) es una enfermedad congénita muy rara, caracterizada por RCIU y posnatal severo (dwarfismo), microcefalia severa, perfil “cabeza de pájaro”, barbilla y frente hacia atrás, nariz grande y picuda, retardo mental severo y otras anomalías. Es un dwarfismo primordial microcefálico: enanismo proporcional extremadamente pequeño para su edad evidente in utero.
Fue descrita por primera vez como “enanismo de cabeza de pájaro” por Rudolf Virchow en 1892. Posteriormente en 1960 Helmut Seckel caracterizó el síndrome tal como lo conocemos en la actualidad, (Bird-Headed Dwarfs. Studies in developmental anthropology including human proportions. Springfield, 111. 1960 Charles C. Thomas, Pu-blishers). Es debido a mutaciones genéticas en los cromosomas 3, 18 y 14.
SCKL es un raro tipo de dwarfismo primordial heterogéneo de prevalencia desconocida. Más de 100 casos a la fecha han sido reportados. En EE.UU y Canadá se estima 1 en 3 millones; en UK 1 en 5 millones. Afecta por igual a mujeres y hombres, sin que parezca presentar predominio étnico ni geográfico. Se transmite como herencia autosómica recesiva y posee heterogenicidad genética: SCKL1. Chromosome 3q22.1-q24 Pakistani families; SCKL2. Chromosome 18q11.31 q1 Iraqi family; SCKL3. Chromosome 14q23 Turkish families; SCKL4. Chromosome 21q 22.3 Middle East families.
Características clínicas. Crecimiento: Estatura corta proporcional severa de inicio prenatal; RCIU/Dwarfismo de PBN; retardo del crecimiento posnatal severo. Rasgos característicos: Craneofa-ciales: microcefalia severa, nariz picuda y prominente (“pico-corno”), frente hundida (hacia atrás), ojos anormalmente grandes, cara estrecha, orejas bajas displásicas con lóbulo hipoplásico o ausente, paladar ojival o hendido, micrognatia. Extremidades:clinodactilia del 5o dedo, microdactilia de uno o más dedos (5o). Se acompaña de grados variables de retraso mental. Con menos frecuencia: craneosinostosis, asimetría facial, fisuras palpebrales antimongoloides, estrabismo, malposición dental, hipoplasia del esmalte dental, retraso de la maduración ósea, hipoplasia de los huesos del carpo, luxación radial/hipoplasia del radio proximal, luxación/displasia del desarrollo de la cadera, hipoplasia del peroné proximal, escoliosis, pliegue simiesco, anomalías de los dermatoglifos, cabello escaso, criptorquidia, clitoromegalia. Otras anomalías asociadas: Oculares: astigmatismo y miopía severa; degeneración retineal bilateral de inicio temprano, severa; hipotelorismo; ptosis bilateral; microftalmos; megacórnea; glaucoma; membrana retrolental; coloboma macular; hipoplasia óptica; luxación del cristalino.Esqueléticas: sólo 11 costillas; pie bot: pie plano; rótula hipoplásica. Anomalías del SNC: cerebro pequeño con patrón circunvolucional antropomorfo, simplificado (micrencefalia pongidoide); disgenesia de la corteza cerebral; agenesia del cuerpo calloso; quiste cerebral dorsal; quiste aracnoideo; ventrículos dilatados; paquigiria; agiria; aneurisma intracraneal. Anomalías endocrinas: anomalías de la glándula pituitaria. Inmunodeficiencia y predisposición a cánceres.
Los métodos diagnósticos se realizan a través de USG prenatal, características clínicas, hallazgos radiológicos, edad ósea retardada, displasia de la cadera, luxación de la cabeza del radio. El diagnóstico diferencial se hace con los dwarfismos primordiales: Majewski Osteodysplastic Primordial Dwarfism Type I (MOPD I); Majewski Osteodysplastic Primordial Dwarfism Type II (MOPD II); Majewski Osteodysplastic Primordial Dwarfism Type III (MOPD III); Russell Silver Syndrome; Meier-Gorlin Syndrome.
Historia natural. La gestación puede prolongarse, el peso al nacer es entre 450 a 1600 g, con un promedio de 1100 a 1300 g. La talla 34 a 43 cm, y talla final post puberal es menos de 100 cm. El P.C.: 22 a 29 cm; con P.C. final post puberal: 38.5 a 41 cm. Hay déficit mental moderado a severo, aunque el progreso motor temprano pueda estar cerca de lo normal. El cerebro es pequeño con patrón de circunvoluciones primitivas simples parecido a un chimpancé. Tienden a ser amistosos y agradables. Son a menudo hiperquinéticos y fácilmente distraídos. Pobre soporte y desarrollo de las articulaciones como luxación de cadera, codo y desarrollo posterior de escoliosis, cifosis. Se ha reportado una sobrevida hasta de 75 años.
Manejo y tratamiento. Correcciones esqueléticas; correcciones oftalmológicas. Anomalías hematológicas: anemia, pancitopenia, leucemia mieloide aguda. Retardo mental. Consejería genética ya que aumenta la frecuencia en padres consanguíneos, riesgo de recurrencia de un 25%; en otros miembros de la familia es baja la frecuencia, a menos que sean consanguíneos.
Fue descrita por primera vez como “enanismo de cabeza de pájaro” por Rudolf Virchow en 1892. Posteriormente en 1960 Helmut Seckel caracterizó el síndrome tal como lo conocemos en la actualidad, (Bird-Headed Dwarfs. Studies in developmental anthropology including human proportions. Springfield, 111. 1960 Charles C. Thomas, Pu-blishers). Es debido a mutaciones genéticas en los cromosomas 3, 18 y 14.
SCKL es un raro tipo de dwarfismo primordial heterogéneo de prevalencia desconocida. Más de 100 casos a la fecha han sido reportados. En EE.UU y Canadá se estima 1 en 3 millones; en UK 1 en 5 millones. Afecta por igual a mujeres y hombres, sin que parezca presentar predominio étnico ni geográfico. Se transmite como herencia autosómica recesiva y posee heterogenicidad genética: SCKL1. Chromosome 3q22.1-q24 Pakistani families; SCKL2. Chromosome 18q11.31 q1 Iraqi family; SCKL3. Chromosome 14q23 Turkish families; SCKL4. Chromosome 21q 22.3 Middle East families.
Características clínicas. Crecimiento: Estatura corta proporcional severa de inicio prenatal; RCIU/Dwarfismo de PBN; retardo del crecimiento posnatal severo. Rasgos característicos: Craneofa-ciales: microcefalia severa, nariz picuda y prominente (“pico-corno”), frente hundida (hacia atrás), ojos anormalmente grandes, cara estrecha, orejas bajas displásicas con lóbulo hipoplásico o ausente, paladar ojival o hendido, micrognatia. Extremidades:clinodactilia del 5o dedo, microdactilia de uno o más dedos (5o). Se acompaña de grados variables de retraso mental. Con menos frecuencia: craneosinostosis, asimetría facial, fisuras palpebrales antimongoloides, estrabismo, malposición dental, hipoplasia del esmalte dental, retraso de la maduración ósea, hipoplasia de los huesos del carpo, luxación radial/hipoplasia del radio proximal, luxación/displasia del desarrollo de la cadera, hipoplasia del peroné proximal, escoliosis, pliegue simiesco, anomalías de los dermatoglifos, cabello escaso, criptorquidia, clitoromegalia. Otras anomalías asociadas: Oculares: astigmatismo y miopía severa; degeneración retineal bilateral de inicio temprano, severa; hipotelorismo; ptosis bilateral; microftalmos; megacórnea; glaucoma; membrana retrolental; coloboma macular; hipoplasia óptica; luxación del cristalino.Esqueléticas: sólo 11 costillas; pie bot: pie plano; rótula hipoplásica. Anomalías del SNC: cerebro pequeño con patrón circunvolucional antropomorfo, simplificado (micrencefalia pongidoide); disgenesia de la corteza cerebral; agenesia del cuerpo calloso; quiste cerebral dorsal; quiste aracnoideo; ventrículos dilatados; paquigiria; agiria; aneurisma intracraneal. Anomalías endocrinas: anomalías de la glándula pituitaria. Inmunodeficiencia y predisposición a cánceres.
Los métodos diagnósticos se realizan a través de USG prenatal, características clínicas, hallazgos radiológicos, edad ósea retardada, displasia de la cadera, luxación de la cabeza del radio. El diagnóstico diferencial se hace con los dwarfismos primordiales: Majewski Osteodysplastic Primordial Dwarfism Type I (MOPD I); Majewski Osteodysplastic Primordial Dwarfism Type II (MOPD II); Majewski Osteodysplastic Primordial Dwarfism Type III (MOPD III); Russell Silver Syndrome; Meier-Gorlin Syndrome.
Historia natural. La gestación puede prolongarse, el peso al nacer es entre 450 a 1600 g, con un promedio de 1100 a 1300 g. La talla 34 a 43 cm, y talla final post puberal es menos de 100 cm. El P.C.: 22 a 29 cm; con P.C. final post puberal: 38.5 a 41 cm. Hay déficit mental moderado a severo, aunque el progreso motor temprano pueda estar cerca de lo normal. El cerebro es pequeño con patrón de circunvoluciones primitivas simples parecido a un chimpancé. Tienden a ser amistosos y agradables. Son a menudo hiperquinéticos y fácilmente distraídos. Pobre soporte y desarrollo de las articulaciones como luxación de cadera, codo y desarrollo posterior de escoliosis, cifosis. Se ha reportado una sobrevida hasta de 75 años.
Manejo y tratamiento. Correcciones esqueléticas; correcciones oftalmológicas. Anomalías hematológicas: anemia, pancitopenia, leucemia mieloide aguda. Retardo mental. Consejería genética ya que aumenta la frecuencia en padres consanguíneos, riesgo de recurrencia de un 25%; en otros miembros de la familia es baja la frecuencia, a menos que sean consanguíneos.

Comentarios
Publicar un comentario