Sindrome de Cornelia de Lange
El síndrome de Cornelia de Lange (siglas en inglés CdLS) es una alteración genética poco conocida que conduce a anormalidades severas del desarrollo. Afecta tanto al desarrollo físico como intelectual del niño. La incidencia exacta se desconoce, estimándose alrededor de 1 entre 10.000 a 30.000.




En 1933, una pediatra holandesa, la Dra. Cornelia de Lange, describía dos niños con rasgos similares. Hoy en día es ella a quien se le reconoce el haber descrito los síntomas que abarcan el síndrome que lleva su nombre. El síndrome también ha sido llamado Brachmann-de Lange, ya que fue el Dr. W. Brachmann quien describió en 1916 síntomas similares en un paciente. Dicho síndrome es congénito, es decir que está presente desde el nacimiento. Aunque haya personas que, aunque lo tengan, no presentan todos los signos y síntomas, sino que tendrán los suficientes como para ser diagnosticados.
Las características más comunes incluyen bajo peso al nacer (usualmente, pero no siempre, por debajo de 2.200 g aprox), crecimiento retardado, baja estatura y cabeza pequeña (microcefalia).
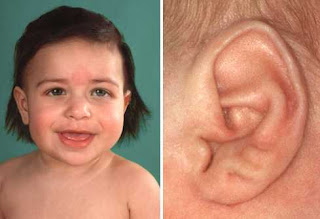
Características faciales típicas incluyen cejas (suelen unirse en el medio) y pestañas muy largas, nariz pequeña y respingona y labios finos en V invertida.
Otras características frecuentes incluyen excesivo pelo en el cuerpo (hirsutismo), manos y pies pequeños, unión parcial del segundo y tercer dedos de los pies, 5º dedo (meñique) curvado, reflujo gastroesofágico, convulsiones, defectos cardíacos, fisura del paladar, anormalidades intestinales, dificultad a la hora de alimentarlos, "incapacidad para prosperar", perdida de audición y vista, y retraso en el desarrollo.
Las anormalidades de los miembros consisten, a veces, en la ausencia de la parte superior en uno o más de ellos (focomelia) o la falta de un dedo o más de las manos o de los pies (oligodactilia). Se encuentran en los casos más severamente afectados.
Existe en algunos pacientes limitación a la extensión de los codos, también puede presentar en varones criptorquidia(genitales pequeños) y llanto de tono bajo y en algunos casos ombligo pequeño.

Existen tres tipos de Síndrome de Cornelia de Lange que varían de grados leves a grados graves
- El tipo 1 o forma clásica: los pacientes presentan restricción de crecimiento intrauterino (RCIU), retardo psicomotor de moderado a profundo y malformaciones mayores que causan discapacidades severas o la muerte.
- El tipo 2 o forma más leve: los signos faciales son menos acusados o se presentan más tardíamente, el retraso mental es leve o no existe, el daño neurológico es leve y los problemas del comportamiento son suaves o inexistentes. En el tipo 2 las malformaciones de los miembros y otras posiblemente acompañantes son menos frecuentes y graves.
- El tipo 3 o fenocopia muestra una gran variabilidad clínica, los rasgos faciales son similares pero la expresión es parcial y con frecuencia se aprecian anomalías cromosómicas. El diagnóstico prenatal es posible en algún hermano de un caso conocido, cuando se aprecian anomalías de los miembros, retraso del crecimiento y microcefalia mediante ultrasonografía.
- Causas del síndrome Cornelia de LangeGenéticasLa mayoría de los casos son debidos a mutaciones espontáneas, aunque los genes afectados causantes de la enfermedad pueden ser heredados de ambos progenitores, haciéndolo de forma autosómica dominante.
En 2004, investigadores del Children’s Hospital of Philadelphia descubrieron un gen cuya mutación es responsable del síndrome (gen NIPBL localizado en la región 5p13-14 del cromosoma 5), y que causa alrededor la mitad de los casos de Cornelia de Lange. En 2006, fue encontrado un segundo gen (SMC1A en la región Xp11.2 del cromosoma X), por científicos italianos. El mismo equipo de investigación de Filadelfia que descubrió el primero, halló un tercer gen en 2007, el gen SMC3 en la región 10q25 del cromosoma 10. Estos dos últimos genes parecen correlacionarse con una forma más suave del síndrome.
Frecuencia
Incidencia: 1/10.000 - 1/20.000 recién nacido. Se estima que, en España, la prevalencia es de 0,97 por 100.000 niño/as nacidos/as vivos/as. No existe predominio en cuanto al sexo.
Expectativa de vida
La esperanza de vida de las personas que padece dicho síndrome no se puede saber a ciencia cierta. Anteriormente, muchos niños fallecieron debido a problemas médicos serios en la infancia por no detectar de manera anticipada sus necesidades. Hoy en día esto no ocurre ya que se espera que la mayoría de ellos vivan hasta bien entrada su adultez. Actualmente pueden vivir mucho tiempo en el ambiente familiar proporcionándoles buenas terapias y una buena atención médica, obteniendo grandes logros.
Patologías asociadas
En el periodo de recién nacido y lactante la dificultad para la alimentación es muy frecuente y habrá que tratarlo tempranamente con métodos médicos habituales y tratamiento quirúrgico. La patología respiratoria ocurre en el 25% de los casos, alguna vez como resultado del reflujo. La patología oftalmológica es muy frecuente que aparezcan estos déficit: miopía, estrabismo y nistagmos, pero el tratamiento es más dificultoso porque los niños no soportan las gafas. Respecto a la sordera de grado variable que aparece en el 20% de lo casos, el diagnóstico y tratamiento que se debe llevar a cabo debe ser precoz pues el retardo del lenguaje puede estar condicionado por ella. Las malformaciones cardíacas se presentan en el 25% de los casos con una gran variabilidad. Generalmente, las personas con este síndrome tienen cierto grado de retraso mental que va de leve a profundo. En la mayoría de los casos los niños con el Síndrome Cornelia de Lange tienen un retraso que va desde leve a moderado. También, estas personas tienen problemas de comportamiento que se determina por: poca habilidad en las relaciones sociales, comportamiento estereotipado y poca expresión facial de las emociones. En pocas ocasiones se ha comprobado un comportamiento autoagresivo, pero incluso en los individuos con tipo 2 (forma más leve) del síndrome existe un perfil de personalidad rígido y muy de acuerdo con un medio ambiente muy ordenado.


Comentarios
Publicar un comentario