Intersex
Intersex, in humans and other animals, is a
variation in sex characteristics including chromosomes, gonads, and/or genitals
that do not allow an individual to be distinctly identified as male or female.
Such variation may involve genital ambiguity, and combinations of chromosomal
genotype and sexual phenotype other than XY-male and XX-female.

Intersex infants with ambiguous outer genitalia
may be surgically 'corrected' to more easily fit into a socially accepted sex
category. Others may opt, in adulthood, for surgical procedures in order to
align their physical sex characteristics with their gender identity or the sex
category to which they were assigned at birth. Others will not become aware
that they are intersex—unless they receive genetic testing—because it does not
manifest in their phenotype. Some individuals may be raised as a certain sex
(male or female) but then identify with another later in life, while others may
not identify themselves as either exclusively female or exclusively male.Research
has shown gender identity of intersex individuals to be independent of sexual
orientation, though some intersex conditions also affect an individual's sexual
orientation.
Intersexuality as a term was adopted by medicine
during the 20th century. Intersex conditions received attention from intersex
activists, who criticized traditional medical approaches in sex assignment and
sought to be heard in the construction of new approaches.The passports and
identification documents of some nationalities have adopted "X" as a
valid third category besides "M" (male) and "F" (female).
Research in the late 20th century has led to a
growing medical consensus that diverse intersex bodies are normal—if relatively
rare—forms of human biology. Milton Diamond, one of the most outspoken experts
on matters affecting intersex people, stresses the importance of care in the
selection of language related to such people.
In humans, biological sex is determined by five
factors present at birth:
- the number and type of sex chromosomes;
- the type of gonads—ovaries or testicles;
- the sex hormones,
- the internal reproductive anatomy (such as the
uterus in females), and
- the external genitalia.
.jpg)
People whose five characteristics are not
either all typically male or all typically female are intersexed.
Conditions and scope
There are a variety of opinions on what
conditions are and are not intersex. For instance, the defunct Intersex Society
of North America (ISNA) definition states that the following conditions
"sometimes involve intersex anatomy" (note this does not mean they
are always intersex conditions):
- 5-alpha reductase deficiency
- androgen insensitivity syndrome
- aphallia
- clitoromegaly
- congenital adrenal hyperplasia
- gonadal dysgenesis (partial &
complete)
- hypospadias
- Klinefelter syndrome
- micropenis
- mosaicism involving sex chromosomes
- ovo-testes (formerly called "true
hermaphroditism")
- partial androgen insensitivity syndrome
- progestin-induced virilisation
- Swyer syndrome
- Turner syndrome
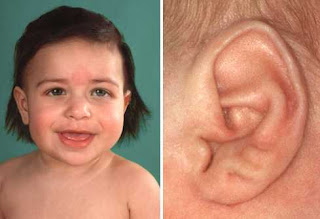
Cryptorchidism
.jpg)
Prevalence
The prevalence of intersex depends on which
definition is used. According to the ISNA definition above, 1 percent of live
births exhibit some degree of sexual ambiguity.[9] Between 0.1% and 0.2% of
live births are ambiguous enough to become the subject of specialist medical
attention, including surgery to assign them to a given sex category (i.e. male
or female). According to Fausto-Sterling's definition of intersex, on the
other hand, 1.9 percent of human births are intersex.
According to Leonard Sax the prevalence of
intersex "restricted to those conditions in which chromosomal sex is inconsistent
with phenotypic sex, or in which the phenotype is not classifiable as either
male or female" is about 0.018%.
Signs
There are a range of variations between female
and male types of genitalia; the Prader scale demonstrates this and is well
illustrated here.
.jpg)
Ambiguous genitalia
Ambiguous genitalia appear as a large clitoris
or small penis.

Because there is variation in all of the
processes of the development of the sex organs, a child can be born with a
sexual anatomy that is typically female, or feminine in appearance with a
larger-than-average clitoris (clitoral hypertrophy), or typically male,
masculine in appearance with a smaller-than-average penis that is open along
the underside. The appearance may be quite ambiguous, describable as female
genitals with a very large clitoris and partially fused labia, or as male
genitals with a very small penis, completely open along the midline
("hypospadic"), and empty scrotum.
Fertility is variable. According to some, the
distinctions "male pseudohermaphrodite", "female
pseudohermaphrodite" and especially "true hermaphrodite"[16] are
vestiges of outdated 19th century thinking. According to others, the terms
"male pseudohermaphrodite", and "female
pseudohermaphrodite" are used to define the gender in terms of the
histology (microscopic appearance) of the gonads.
"True hermaphroditism"
A "true hermaphrodite" is defined as
someone with both testicular and ovarian tissue.

In 2003, researchers at UCLA published their
studies of a lateral gynandromorphic hermaphroditic zebra finch, which had a
testicle on the right and an ovary on the left. Its entire body was split down
the middle between female and male, with hormones from both gonads running
through the blood.This is an example of mosaicism or chimerism.
This extreme example of hermaphroditism is
quite rare.
Ovotestes
Though naturally occurring true hermaphroditism
in humans is unknown, there is, on the other hand, a spectrum of forms of
ovotestes. The varieties include having two ovotestes or one ovary and one
ovotestis, often in the form of streak gonads. Phenotype is not determinable
from the ovotestes; in some cases, the appearance is "fairly typically
female"; in others, it is "fairly typically male," and it may
also be "fairly in-between in terms of genital development.
Intersex activist Cheryl Chase is an example of
someone with ovotestes.

Other diagnostic signs
In order to help in classification, methods
other than a genitalia inspection can be performed:
For instance, a karyotype display of a tissue
sample may determine which of the causes of intersex is prevalent in the case.
Management
Clinical management of intersex can be
categorized into one of the following two:
Treatments: Restore functionality (or potential
functionality)
Enhancements: Give the ability to identify with
“mainstream” people, e.g., breast enlargement surgery
However, there are other categorization systems
of management of intersex, which falls into neither category.
In any case, the most common procedure is
surgery.
Surgery

The exact procedure of the surgery depends on
what is the cause of a less common body phenotype in the first place. There is
often concern as to whether surgery should be performed at all. A traditional
approach to the management of intersexuality has been surgery. However,
some such as Alice Dreger say that surgical treatment is socially motivated
and, hence, ethically questionable; without evidence, doctors regularly assume
that intersex persons cannot have a clear gender identity. This is often taken
further with parents of intersex babies advised that without surgery their
child will be stigmatized. Further, since almost all such surgeries are
undertaken to fashion female genitalia for the child, it is more difficult for
the child to present as male if they later find they identify as or are genetically
male. 20-50% of surgical cases result in a loss of sexual sensation (Newman
1991, 1992).
Typically, surgery is performed at birth.
Intersex advocates such as Anne Fausto-Sterling in her Sexing the Body argue
surgery on intersex babies should wait until the child can make an informed
decision, and label surgery without consent as genital mutilation.
Causes
Typical sex development
The common pathway of sexual differentiation,
where a productive human female has an XX chromosome pair, and a productive
male has an XY pair, is relevant to the development of intersex conditions.

During fertilization, the sperm adds either an
X (female) or a Y (male) chromosome to the X in the ovum. This determines the
genetic sex of the embryo.During the first weeks of development, genetic male
and female fetuses are "anatomically indistinguishable," with
primitive gonads beginning to develop during approximately the sixth week of
gestation. The gonads, in a "bipotential state," may develop into
either testes (the male gonads) or ovaries (the female gonads), depending on
the consequent events.Through the seventh week, genetically female and genetically
male fetuses appear identical.
At around eight weeks of gestation, the gonads
of an XY embryo differentiate into functional testes, secreting testosterone.
Ovarian differentiation, for XX embryos, does not occur until approximately
Week 12 of gestation. In normal female differentiation, the Müllerian duct
system develops into the uterus, Fallopian tubes, and inner third of the
vagina. In males, the Müllerian duct-inhibiting hormone MIH causes this duct
system to regress. Next, androgens cause the development of the Wolffian duct
system, which develops into the vas deferens, seminal vesicles, and ejaculatory
ducts.By birth, the typical fetus has been completely "sexed" male or
female, meaning that the genetic sex (XY-male or XX-female) corresponds with
the phenotypical sex; that is to say, genetic sex corresponds with internal and
external gonads, and external appearance of the genitals.
Conditions
This section needs additional citations for
verification. Please help improve this article by adding citations to reliable
sources. Unsourced material may be challenged and removed. (April 2010)
The final body appearance does not always
correspond with what is dictated by the genes. In other words, there is
sometimes an incongruity between genetic (or chromosomal) and phenotypic (or
physical appearance) sex. Citing medical research regarding other factors that
influence sexual differentiation, the Intersex Society of North America
challenges the XY sex-determination system's assumption that chromosomal sex is
the determining factor of a person's "true" biological sex.
XX Congenital adrenal hyperplasia (CAH) The
most common cause of sexual ambiguity is congenital adrenal hyperplasia (CAH),
an endocrine disorder in which the adrenal glands produce abnormally high
levels of virilizing hormones in utero

In XX-females, this can range from partial
masculinization that produces a large clitoris, to virilization and male
appearance. The latter applies in particular to Congenital adrenal hyperplasia
due to 21-hydroxylase deficiency, which is the most common form of CAH.
Individuals born with XX chromosomes affected
by 17α-hydroxylase deficiency are born with female internal and external
anatomy, but, at puberty, neither the adrenals nor the ovaries can produce
sex-hormones, inhibiting breast development and the growth of pubic hair.
XX Progestin-induced virilisation In this case, the excess androgen hormones are caused by use of
progestin, a drug that was used in the 1950s and 1960s to prevent miscarriage.
These individuals normally have internal and external female anatomy, with
functional ovaries and will therefore have menstruation. They develop, however,
some male secondary sex characteristics and they frequently have unusually
large clitorises. In very advanced cases, such children have initially been
identified as males.

XX Freemartinism This
condition occurs commonly in all species of cattle and affects most females
born as a twin to a male. It is rare or unknown in other mammals, including
humans. In cattle, the placentae of fraternal twins usually fuse at some time
during the pregnancy, and the twins then share their blood supply. If the twins
are of different sexes, male hormones produced in the body of the fetal bull
find their way into the body of the fetal heifer (female), and masculinize her.
Her sexual organs do not develop fully, and her ovaries may even contain
testicular tissue. When adult, such a freemartin is very like a normal female
in external appearance, but she is infertile, and behaves more like a castrated
male (a steer). The male twin is not significantly affected, although (if he
remains entire) his testes may be slightly reduced in size. The degree of
masculinization of the freemartin depends on the stage of pregnancy at which
the placental fusion occurs – in about ten percent of such births no fusion
occurs and both calves develop normally as in other mammals.

XY Androgen insensitivity syndrome (AIS) People with AIS have a Y chromosome, (typically
XY), but are unable to metabolize androgens in varying degrees.
Cases with typically female appearance and
genitalia are said to have complete androgen insensitivity syndrome (CAIS).
People with CAIS have a vagina and no uterus, cervix, or ovaries, and are
infertile. The vagina may be shorter than usual, and, in some cases, is nearly
absent. Instead of female internal reproductive organs, a person with CAIS has
undescended or partially descended testes, of which the person may not even be
aware.

In mild and partial androgen insensitivity
syndrome (MAIS and PAIS), the body is partially receptive to androgens, so
there is virilization to varying degrees. PAIS can result in genital ambiguity,
due to limited metabolization of the androgens produced by the testes.
Ambiguous genitalia may present as a large clitoris, known as clitoromegaly, or
a small penis, which is called micropenis or microphallus; hypospadias and
cryptorchidism may also be present, with one or both testes undescended, and
hypospadias appearing just below the glans on an otherwise typical male penis,
or at the base of the shaft, or at the perineum and including a bifid (or
cleft) scrotum.
XY 5-alpha-reductase deficiency (5-ARD) The condition affects individuals with a Y
chromosome, making their bodies unable to convert testosterone to
dihydrotestosterone (DHT). DHT is necessary for the development of male
genitalia in utero, and plays no role in female development, so its absence
tends to result in ambiguous genitalia at birth; the effects can range from
infertility with male genitalia to male underdevelopment with hypospadias to
female genitalia with mild clitoromegaly.

The frequency is unknown, and
children are sometimes misdiagnosed as having AIS. Individuals can have
testes, as well as vagina and labia, and a small penis capable of ejaculation
that looks like a clitoris at birth. Such individuals are usually raised as
girls. The lack of DHT also limits the development of facial hair.
XY Congenital adrenal hyperplasia (CAH) In individuals with a Y chromosome (typically XY) who have Congenital
adrenal hyperplasia due to 17 alpha-hydroxylase deficiency, CAH inhibits
virilization, unlike cases without a Y chromosome.

XY Persistent Müllerian duct syndrome (PMDS) The
child has XY chromosomes typical of a male. The child has a male body and an
internal uterus and fallopian tubes because his body did not produce Müllerian
inhibiting factor during fetal development.

XY Anorchia Individuals
with XY chromosomes whose gonads were lost after 14 weeks of fetal development.
People with Anorchia have no ability to produce the hormones responsible for
developing male secondary sex characteristics nor the means to produce gametes
necessary for reproduction due to the lack of gonads.

They may develop
typically feminine secondary sex characteristics without or despite the
administration of androgens to artificially initiate physical sex
differentiation (typically planned around the age of puberty). Psychological
and neurological gender identity may solidify before the administration of
androgens, leading to gender dysphoria, as anorchic individuals are typically
assigned male at birth.
XY Gonadal Dysgenesis It
has various causes and are not all genetic; a catch-all category.
It refers to individuals (mostly XY) whos
gonads don't develop properly.

Clinical features are heterogeneous.
XY Hypospadias It
is caused by various causes,including alterations in testosterone metabolism.
The urethra does not run to the tip of the
penis. In mild forms, the opening is just shy of the tip; in moderate forms, it
is along the shaft; and in severe forms, it may open at the base of the penis.

Other:Unusual
chromosomal sex
In addition to the
most common XX and XY chromosomal sexes, there are several other possible
combinations, for example Turner syndrome (XO), Triple X syndrome (XXX),
Klinefelter's Syndrome, (XXY/XXXY), XYY syndrome (XYY), de la Chapelle syndrome
(XX male), Swyer syndrome (XY female), and there are many other individuals who
do not follow the typical patterns (such as individuals with four or even more
sex chromosomes).

Klinefelter's Syndrome
Other: Mosaicism
and chimerism
A mix can occur,
where some of the cells of the body have the common XX or XY, while some have
one of the less usual chromosomal contents above. Such a mixture is caused by
either mosaicism or chimerism. In mosaicism, the mixture is caused by a
mutation in one of the cells of the embryo after fertilization, whereas
chimerism is a fusion of two embryos.

Mosaicism
In alternative fashion, it is simply a mixture
between XX and XY, and does not have to involve any less-common genotypes in
individual cells. This, too, can occur both as chimerism and as a result of one
sex chromosome having mutated into the other.
However, not all cases of mosaicism and
chimerism involve intersex.
Complications
In the cases where nonfunctional testes are
present, there is a risk that these develop cancer. Therefore, doctors either
remove them by orchidectomy or monitor them carefully. This is the case for
instance in androgen insensitivity syndrome.

Identification
documents
Nepal, the United Kingdom, Australia, Portugal,
Uruguay, and New Zealand are states that took steps to allow for transgender
and intersex people to list their gender as other than male or female.[30]
Australian and New Zealand authorities allow passports to have a third gender
option which is marked "X" meaning
"indeterminate/unspecified/intersex" instead of "F" for
female or "M" for male. The move was seen as removing discrimination
against transgender and intersex people so they will no longer have to choose
between "M" (male) or "F" (female).

Comentarios
Publicar un comentario