Ictiosis Arlequin

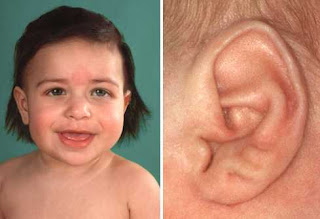
Ictiosis arlequín es una enfermedad de la piel extremadamente rara del grupo de las llamadas genodermatosis (grupo de dermatosis hereditarias con trastornos metabólicos). Es la forma de ictiosis congénita más grave, se hace evidente ya desde el nacimiento y debe su nombre al aspecto que tienen los recién nacidos con la enfermedad, que recuerda a un disfraz de arlequín. La ictiosis tipo Arlequín es una enfermedad genética rara de la piel caracterizada por escamas grandes y gruesas que aparecen en toda la piel, como a su vez se nace con los párpados volteados por lo que en lugar de ojos se observan los párpados totalmente rojos. Se asocia generalmente deformidades faciales características y a menudo anomalías en otras partes del cuerpo, especialmente en el tórax.
Se debe a una alteración de la queratinización cuyo mecanismo fisiopatológico se desconoce, pero se piensa que existe una disgenesia de la capa lamelar, probablemente debida a anomalías de los lípidos cutáneos, que da lugar a una hiperqueratosis folicular masiva.
Aparecen fisuras profundas e irregulares que cubren la superficie corporal, ectropión (eversión de los párpados) y eclabium (inversión hacia fuera de los labios) debidos a la tracción mecánica que ejerce la piel engrosada sobre la conjuntiva y la mucosa oral, hipoplasia (desarrollo incompleto o defectuoso) de orejas nariz y dedos y los recién nacidos adoptan una postura semiflexionada.
Las complicaciones clínicas más importantes se producen a causa del fallo de la función de barrera que la piel tiene en condiciones normales e incluyen sepsis (infección o contaminación generalizada) y deshidratación que conducen a hipernatremia (aumento anormal de sodio en sangre) y malnutrición.
Los niños nacen con constricción marcada de tórax y abdomen con las correspondientes dificultades respiratorias y de alimentación.
El pronóstico es malo, ya que la mayoría fallece a los pocos días o semanas del nacimiento. Aunque existen casos excepcionales en donde la persona llega a la adolescencia, pero necesita de tratamientos especiales.

La base patológica radica en una lesión focal del sistema nervioso simpático encargado de la inervación sudomotora y vasoconstrictora de la cara. Inicialmente el paciente experimenta rubor facial secundario al aumento del tono vasoconstrictor pero poco después, por un mecanismo de compensación, el área facial no denervada se vuele roja y sudorosa mientras que el área denervada se mantiene sin cambios. A pesar de que la mayoría de los casos son idiopáticos, se hace necesario descartar una lesión a nivel de cualquiera de las tres neuronas que forman el simpático cervical:
Neurona de primer orden o central: nace del hipotálamo posterior, discurre por el tegmento del troncoencéfalo para terminar penetrando en la médula espinal donde, a nivel de la columna intermediolateral y a la altura de C8-T2 (centro cilioespinal de Budge) hace sinapsis con la segunda neurona.
Neurona de segundo orden o preganglionar: abandona la médula a través de las raíces C8-T2 formando parte del ganglio estrellado y posteriormente de la cadena simpática paravertebral (en estrecha relación con el ápex pulmonar) que asciende y termina en el ganglio cervical superior, situado a nivel de la bifurcación carotídea. Del ganglio estrellado parten las fibras que inervan a cuello, brazo y parte superior de tronco.
Neurona de tercer orden o postganglionar: del ganglio cervical superior parten dos vías diferentes, donde una se encuentra junto a la carótida interna y está compuesta por las fibras sudomotoras y vasoconstrictoras hacia la frente-nariz, junto a las fibras oculosimpáticas que inervan al músculo dilatador del iris. La otra junto a la carótida externa compuesta de fibras sudomotoras y vasoconstrictorias hacia el resto de la cara, acompañando a las ramas terminales trigeminales. Lesiones de la neurona de tercer orden producirán síntomas autonómicos en frente y lesiones de la neurona de primer o segundo orden en toda la cara.
Las ictiosis pueden ser hereditarias y adquiridas:
- Las ictiosis hereditarias son raras y por lo general aparecen durante la infancia y se mantienen de por vida, siendo frecuentemente formas de afectación leve y que tienden a mejorar durante los meses de verano, aunque también pueden existir algunas formas clínicas muy severas. Las ictiosis hereditarias se caracterizan por el gran acumulo de escamas sobre la superficie cutánea y se clasifican en función de criterios genéticos y clínicos.
- Las ictiosis adquiridas aparecen asociadas a otras enfermedades, sobre todo renales y pueden comenzar en cualquier época de la vida, pueden constituir una manifestación precoz de algunas enfermedades sistémicas (lepra, hipotiroidismo, linfoma, SIDA). La descamación seca puede ser fina y localizarse exclusivamente en el tronco y las piernas, o ser gruesa y difusa. La biopsia (operación que consiste en extirpar en el individuo vivo un fragmento de órgano o de tumor con objeto de someterlo a examen microscópico) de la piel ictiósica no suele resultar diagnóstica, aunque existen excepciones, como la sarcoidosis, en la que se producen escamas gruesas en las piernas y presencia de granulomas típicos en la biopsia.
Aunque la presentación clínica sea la misma, hallazgos histológicos y de laboratorio permiten diferenciar 3 tipos de alteraciones en la morfología de los queratocitos (células productoras de queratina) y la expresión proteica de los mismos.
Es posible el diagnóstico prenatal mediante ecografía, fetoscopia (procedimiento mediante el cual se puede observar directamente al feto dentro del útero, mediante el uso de un fetoscopio introducido a través de una incisión bajo anestesia local) y amniocentesis (procedimiento obstétrico mediante el cual se extrae una pequeña cantidad de liquido amniótico para su posterior análisis).
Aunque se ha utilizado la biopsia de piel fetal, realizada mediante control ecográfico, ésta es una técnica no exenta de riesgos que solamente se debe realizar en centros muy especializados y teniendo en cuenta la posibilidad de pérdida fetal que podría acontecer en un sujeto sano.
El tratamiento inicial se centra en atenuar el trastorno de la función de barrera de la piel, mediante medidas de hidratación, tratamiento antibiótico ante los signos precoces de infección y medidas de soporte respiratorio y nutricionales.
Entre las medidas de soporte cutáneas, en el tratamiento de cualquier tipo de ictiosis, resulta útil reducir al mínimo los baños, emplear, sólo en los pliegues cutáneos, jabones que no contengan hexaclorofeno dado que los pacientes presentan mayor capacidad de absorción y, por tanto, mayor riesgo de toxicidad, aplicar dos veces al día emolientes, del tipo de la vaselina simple, aceite mineral o lociones conteniendo urea o propilenglicol; esta aplicación debe hacerse sobre todo después del baño mientras la piel está todavía húmeda y los productos se deben dejar retirando el exceso mediante golpecitos con la toalla o mantenerse como mínimo 10 minutos. Otras sustancias útiles incluyen el gel de ácido salicílico al 5%, preparados con vaselina hidrofílica y agua a partes iguales, así como cremas que contengan ácido a-hidroxi láctico, glicólico y/o pirúvico en diversas bases.
El uso precoz de retinoides tópicos y en los casos muy severos por vía oral, parece haber mejorado la supervivencia en alrededor de un año, en los niños que sobreviven la ictiosis parece evolucionar hacia una eritrodermia ictiosiforme congénita no ampollosa.
A diferencia de las otras ictiosis congénitas, el defecto genético no ha sido identificado. Se hereda como un rasgo genético autosómico recesivo.

Comentarios
Publicar un comentario